


FDA ADVERSE EVENTS
FDA-reported
Adverse Events by Lens Type
Learn
what 12 years of data tells us about hydrogels, silicone hydrogels, GPs and decorative
contact lenses.

The FDA receives information about medical device adverse events — including those involving contact lenses — from manufacturers, importers and user facilities through its Medical Device Reporting (MDR) program. The FDA uses these reports to detect medical device problems and to intervene if necessary. Since 1984, the FDA has required manufacturers and importers of medical devices to report all device-related deaths, serious injuries and certain malfunctions.
Despite this requirement, the FDA received only a handful of adverse event reports. A 1986 General Accounting Office study showed that less than 1 percent of device problems occurring in hospitals were reported to the FDA, and the more serious the problem with a device, the less likely it was to be reported.
To address this shortcoming, the Safe Medical Devices Act (SMDA) of 1990 required device "user facilities" to also report device-related adverse events to the FDA. The Medical Device Amendments of 1992 clarified certain terms and established a single reporting standard for device user facilities, manufacturers, importers and distributors. It's not fully known to what extent these actions have improved mandatory adverse-event reporting.
The cumulative MDR data is organized under the Device Experience Network (DEN) and the Manufacturer User Facility and Distributor Experience (MAUDE) databases. The DEN database includes reports received under the mandatory MDR program from 1984 to 1996 and voluntary reports from patients and practitioners up to June 1993. The more recent MAUDE database includes mandatory manufacturer reports since August 1996 and voluntary reports from patients and practitioners since June 1993. A user can search each online database for information on medical devices that may have malfunctioned, caused serious injury or even caused death.
|
TABLE 1 |
|
|
Classification of Reported Adverse Events from 1994 to 2005 |
|
| TYPE OF EVENT REPORTED | NUMBER |
| Microbial keratitis, diagnosed or presumed by practitioner or manufacturer | 360 (3) |
|
Non-severe adverse event
including these subcategories: • Abrasion, some due to faulty lenses — 31 • Inflammation, infection other than MK — 20 � Uveitis — 12 • Toxicity reaction from care regimen, packaging solution or unknown — 9 � Neovascularization, edema or corneal exhaustion — 8 • Contact lens papillary conjunctivitis — 5 • Cutting fingers on package foil — 4 |
114 (6) |
| Lens damage, either torn or defective lens | 58 |
| Uncertain diagnosis including pain, irritation, eye injury � unlikely MK | 49 (3) |
| Sold by unregistered vendor | 3 |
| TOTAL | 584 |
| Note: Figures in parentheses were reportedly sold by an unregistered vendor and were categorized accordingly under MK, non-severe adverse event or uncertain diagnosis. There were also three reports of contact lenses sold by an unregistered vendor but without any specified adverse event. | |
About MAUDE Data
The MAUDE database may reflect recent trends of adverse events by contact lens type. To avoid any misunderstanding, be advised that the medical device reporting system isn't a controlled process, meaning that several sources of confounding variables exist that prevent scientific conclusions. In fact, the FDA states that the MAUDE data is not intended for evaluating rates of adverse events or for comparing adverse event occurrence rates across devices. Consistent with this caveat, I did not calculate incidence rates of adverse events, despite temptation to use available market share data for each contact lens type to do exactly that. Similarly, it wasn't possible to perform statistical analyses to determine whether changes in reporting for particular lens types resulted from chance variation or were statistically significant.
Instead, I retrieved 12 years worth of contact lens-related adverse event reports from the FDA Web site. I categorized the contact-lens related adverse events for each year according to lens type (hydrogel, silicone hydrogel, gas permeable and decorative lenses), the type of adverse event and who reported the incident.
This article summarizes trends in adverse events reporting to the FDA by lens type while relating the findings to possible changes in the contact lens industry and prescribing habits over the same period.
What Constitutes a Reportable Event?
The FDA encourages voluntary adverse event reporting by consumers and practitioners using Form 3500 available online. According to the FDA, Form 3500 "should be used by healthcare professionals and consumers for voluntary reporting of serious adverse events, potential and actual product use errors, and product quality problems associated with the use of FDA-regulated drugs, biologics (including human cells, tissues, and cellular and tissue-based products), medical devices (including in vitro diagnostics), special nutritional products and cosmetics."
There is certainly leeway for interpreting what constitutes an adverse event. For example, the FDA asks consumers to submit a report "If you think you or someone in your family has experienced a serious reaction to a medical product..." Practitioners are told that "An adverse event is any undesirable experience associated with the use of a medical product in a patient." Furthermore, the FDA advises practitioners that the event is "serious" when the patient outcome is death, life-threatening, requires hospitalization, involves disability, causes congenital anomaly or requires intervention to prevent permanent impairment or damage.
In contrast to voluntary reporting, medical device manufacturers are required to report all "MDR reportable events" to the FDA on Form 3500A, also available online. A report is required when a manufacturer becomes aware of information that reasonably suggests that one of its marketed devices has or may have caused or contributed to a death or serious injury, or that it has malfunctioned and that the device or a similar device marketed by the manufacturer would likely cause or contribute to a death or serious injury if the malfunction were to recur.
Contact Lens-related Adverse Events
|
|
|
Figure 1. Number of reported MK cases by year and lens type. |
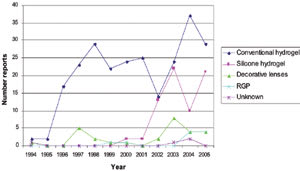
The contact-lens related adverse events I evaluated from the MAUDE database included microbial keratitis (MK), corneal abrasion, unspecified ocular irritation and lens defects such as split lenses and incorrect packaging (Table 1).
About 60 percent (360/584) of the reports were attributed to MK or presumed MK during the 12-year period (Figure 1). MK was the most common reason for an adverse event report. Starting in 1996, an average of 26 reports occurred per year over the next seven years, with hydrogel lenses (defined for the purposes of this article as all hydrogel lens types except for silicone hydrogels and decorative hydrogel lenses) as the predominant lens type. Since 2003, the number of reports attributed to MK increased to more than 50 reports per year.
Nearly 20 percent (114/584) of events were classified as non-severe and included conditions such as corneal neovascularization, papillary conjunctivitis, allergic or toxic response and corneal abrasions. The non-severe events unrelated to contact lenses included idiopathic iritis and four events in which patients cut fingers while handling lens packaging. About 8 percent (49/584) were classified as uncertain diagnoses that were unlikely to be MK. These cases described signs and symptoms but gave no diagnosis, or insufficient information was available to categorize the reported event. About half of the cases of uncertain diagnosis came from a voluntary report on the FDA database, while about one-fifth followed a letter from a patient's attorney to the manufacturer. About 10 percent (58/584) of reports were due to defective lenses, with eight of the 10 categorized as voluntary reporting, many by patients. Reports of defective lenses were fairly consistent in number over the 12-year period, averaging four to five reports per year with the majority (82 percent) associated with hydrogel lenses.
An Increasing Number of Reported Incidents
Between January 1994 and December 2005, a total of 584 contact lens-related adverse events were categorized in the MAUDE database. The annual number of contact lens-related adverse event reports increased from 19 in 1994, when the system was started, to a high of 102 reports in 2003, when approximately one-quarter of the reports involved silicone hydrogels. Since 2003, there has been a slight decline in the number of reports, with 84 in 2005.
The relatively low number of reports in 1994 and 1995 during the inception of the MAUDE database represented voluntary reporting from individual patients or practitioners, often with limited information. Approximately one-third of these early reports were classified as having an uncertain diagnosis because of a lack of information. Since 1996, when manufacturers began recording adverse events in the MAUDE database, the number of reports has steadily risen along with an increased level of detail about each event. The higher volume of reports may also reflect increased patient and practitioner familiarity with the FDA database. Nevertheless, the total of 584 clearly represents a significant under-reporting of all actual cases of adverse contact lens events.
Figure 2 summarizes the results over the 12 years according to lens type. The events were associated with a range of lens types, with hydrogels (403/584) taking the majority, followed by silicone hydrogels (89/584), decorative hydrogels (58/584) and GPs (15/584). Three percent (19/584) of reported incidents involved unknown or miscellaneous lenses, such as Softperm (CIBA Vision).
|
|
|
Figure 2. Reported events by year and lens type. |
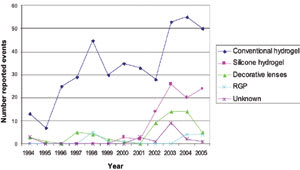
Sources of Reporting
There were no manufacturer-reported events for 1994 or 1995 because the MAUDE database did not tabulate such events until August 1996. Instead, practitioners or patients voluntarily reported the 27 total events in 1994 and 1995. Excluding the MAUDE data from 1994 and 1995, approximately 75 percent (417/557) of the contact lens-related adverse event reports originated through manufacturers (Figure 3) during the time from 1996 to 2005. About 5 percent (29/557) of reports followed communication from a patient's attorney, either directly to the FDA or via the manufacturer or practitioner. Less than 1 percent (5/557) of events came from manufacturers after a case report in a peer-reviewed journal showed that their lens was involved with a significant adverse event. With one-fifth of reports since 1996 reported on a voluntary basis, practitioner or patient bias may have resulted in an under- or over-reporting of events for certain lens types.
Manufacturer reporting of clinical study-related incidents has increased with 15 cases reported since 2000; four of them involving hydrogels and the remaining 11 involving silicone hydrogels. It's possible that other clinical study-related incidents occurred during this same period, but may have escaped identification as coming from a clinical study in the FDA reporting.
It's important to know that the adverse incidents from the MAUDE database are not reports exclusively from the United States. Patients and practitioners worldwide can access the database. Indeed, some manufacturer-generated reports came from a foreign affiliate to a U.S.-based manufacturer that then reported the event. Unfortunately, not enough information is available in the MAUDE data to know how many of the reports originated outside of the United States. Therefore, the trends among the reported incidents may be influenced to some degree by the prescribing in other countries, which we should take into consideration when comparing them to U.S. prescribing trends. It's known that the pattern of contact lens prescribing outside of the United States differs by lens type and modality.
Hydrogel Lenses
The predominant lens type associated with adverse events reported to the FDA has been hydrogels. The number of reported MK events by lens type shows that the occurrence with hydrogels has been fairly static over the last 10 years, averaging 24 reports a year. In 1996, all reports were with hydrogel lenses. However, the proportion of reports related to hydrogels has decreased in recent years, probably because of increased prescribing and manufacturer clinical study of silicone hydrogels. In 2005, hydrogels accounted for 60 percent of reports.
Silicone Hydrogels
|
|
|
Figure 3. Who reported the events (1996-2005) |
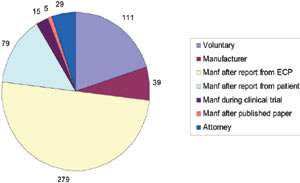
Reported events with silicone hydrogel lenses have increased between 2001 and 2005, with the number of MK reports reasonably similar between silicone hydrogels and hydrogels, except for 2004. Reported MK events with silicone hydrogels have increased over the last four years, with an average of 16 per year. In 2005, there were 21 reports of MK with silicone hydrogels compared to 29 with hydrogels. This increase may have resulted from the growing popularity of silicone hydrogel prescribing and their close monitoring by manufacturers. It's unknown how many MK reports were attributable to daily or extended wear, although industry data shows that practitioners currently prescribe silicone hydrogels more for daily wear. Furthermore, it wasn't always clear how the lenses were worn or prescribed. For example, several reported adverse events involved patients who noncompliantly wore a silicone hydrogel indicated for daily wear overnight.
The first silicone hydrogel lens appeared in 1999 for six-night extended wear, and in 2001 two silicone hydrogels obtained approval for 30-day continuous wear. In the first few years that silicone hydrogel lenses were commercially available, the majority were prescribed for extended wear. The increase in adverse event reporting involving silicone hydrogel lenses results partly from more of the lenses being prescribed, which increased from approximately 1 percent of soft spherical lenses in 2001 to 5 percent in 2003.
After 2003, although the number of reported incidents remained fairly static for silicone hydrogels, the number of these lenses prescribed increased significantly. Among soft spherical lenses, silicone hydrogel lenses accounted for about 18 percent in 2004 and nearly one-third in 2005. During this time, practitioners increasingly prescribed silicone hydrogels for daily wear, with some of the lenses specifically marketed for daily wear. In 2005, approximately two-thirds of silicone hydrogels prescribed had a specific daily wear indication. However, this shift toward daily wear wasn't associated with a decrease in reported events with silicone hydrogels, as we might expect.
You might think that six years following the launch of silicone hydrogels, their increasing use might be associated with fewer reported adverse events compared to hydrogels. However, evaluation of the MAUDE data doesn't support such a hypothesis. A recent study in Australia showed no significant difference in MK incidence with the overnight wear of silicone hydrogels and hydrogels. Other studies have concluded that while overnight wear increases MK incidence, silicone hydrogel overnight wear is associated with less risk than hydrogel overnight wear. Even with more evidence emerging about contact lens safety, it remains unknown whether silicone hydrogels provide greater safety for overnight wear than do hydrogels. This realization should stimulate practitioners to critically assess silicone hydrogel clinical performance for daily wear and extended wear.
While the MAUDE data shows a relatively high number of silicone hydrogel events, it's not possible to determine whether this is abnormal. For example, this finding may result from practitioners prescribing a disproportionate number of silicone hydrogels for problematic or "at-risk" patients. It's also possible that the newness and interest in silicone hydrogels compared to other lens technologies is leading to an over-represented number of reports. Finally, this data may represent extensive clinical study and monitoring of silicone hydrogels by manufacturers compared to other lens types.
Gas Permeable Contact Lenses
As some of you might expect, the number of adverse event reports with GP lenses was low over the 12 years. However, a small increase in reported events occurred in 2004 and 2005; all were cases of MK with overnight wear of high-Dk/t orthokeratology lenses. This appears related to increased practitioner prescribing of overnight corneal reshaping GPs in recent years following the FDA approval of Corneal Refractive Therapy (Paragon Vision Sciences) in 2002; Contex OK-E (Contex) and Emerald (Euclid Systems) in 2004; and Vision Shaping Treatment (Bausch & Lomb) in 2005. Despite evidence for the clinical efficacy of ortho-k for the temporary reduction of myopia, there are increasing reports of MK with ortho-k. Non-compliance and use of tap water are common risk factors. Although there still is no known incidence for MK with overnight ortho-k, several studies have identified that overnight ortho-k is an important risk factor for MK, especially for children.
Decorative Lenses
The steady but low number of adverse events for decorative lenses changed between 2002 and 2004, with a substantial increase in reporting and an average of 12 annual cases in 2003 and 2004. The number of reports compared to their relatively low use might be concerning, although a reduction to just five events followed in 2005. It wasn't possible to differentiate whether the decorative lenses were plano or for correcting refractive error, but approximately one-quarter were allegedly supplied without a prescription by non-licensed vendors. This may have caused the increase in reporting a few years ago, as practitioners may have wanted to demonstrate to the FDA the existence of adverse events including MK with decorative lenses. Unlicensed vendors sold some of these decorative lenses without confirming that the patients had an ocular assessment, customized fitting or verbal counseling regarding lens care, and they offered no provision for aftercare. With several published cases of vision loss related to the improper wear and care of decorative contact lenses, legislators now seem to understand that the potential complications exist whether or not the contact lenses correct for refractive error. Publicity generated in the professional and lay press during this time about the risks associated with wearing plano, decorative lenses without a prescription helped to effect legislative changes, culminated by the passage of a bill in November 2005 to protect patients from eye injuries resulting from inappropriately filled prescriptions for decorative lenses. The reduction in 2005 of reports with decorative lenses may have resulted from the new legislation to protect patients. It's possible that practitioners are satisfied knowing that plano decorative lenses are regulated like all other contact lenses, such that they don't have as much incentive to point out decorative contact lens complications to the FDA.
Conclusions
Although it's unknown how accurately this summary reflects true clinical experience with various contact lenses, it does offer perspective on changes that have taken place in the contact lens industry. For example, the increased reporting of adverse events with decorative lenses in 2003 and 2004 seems related with decorative lens legislation introduced during that time. Additionally, the notable increase in adverse event reporting involving silicone hydrogel lenses follows the dramatic trend of practitioners prescribing these lenses in greater numbers. As a final illustration, the upward trend of events with GPs coincides with increased prescribing of overnight corneal reshaping.
Yet it's difficult to ascertain the diagnoses and lens types reported to the FDA, along with other relevant information such as patient compliance and whether patients wore the lenses on a daily or extended wear basis. With these limitations in mind, monitoring the FDA data may offer an alternative tool for examining contact lens industry trends to supplement the more traditional estimated sales volume and practitioner surveys.
Dr. Chou acknowledges Visioncare Research Ltd. for its expertise in the data compilation and Mark Bullimore, MCOptom, PhD, FAAO, at The Ohio State University College of Optometry, and his vision science students for reviewing this article.
To obtain references for this article, please visit http://www.clspectrum.com/references.asp and click on document #133.
Dr. Chou is in group practice in San Diego at Carmel Mountain Vision Care. He is also the co-developer of EyeDock.com, an online contact lens reference for doctors.