


continuing education
Understanding Microbiology and Contact Lens Solutions
Learn how a greater knowledge of microbiology has helped improve the safety and efficacy of contact lenses, solutions and accessories.
BY WILLIAM J. GLEASON, OD
An article I published in Contact Lens Spectrum in 1999 began with a discussion on the principal objective of the Food and Drug Administration (FDA), which is to provide a reasonable assurance of the safety and efficacy of drugs and devices before and after they're either approved or cleared for public distribution. The article also discussed patient compliance following approval — a major problem in many areas of health care, and contact lenses are no exception.
Ten years later, these discussion points are still valid. FDA approvals can be viewed as only providing reasonable assurance. Despite the views of plantiff lawyers to the contrary, the FDA is unable to evaluate products for absolute safety and efficacy. Absolute safety is unattainable because the potential for problems with contact lenses and solutions will always exist, either through patient misuse or misfortune. However, over the past three decades, the evolution of approval requirements for devices has reduced these problems.
This article looks at how the regulations and clinical controls established by the Food and Drug Administration over the past three decades have changed and evolved to improve the safety and efficacy of contact lenses, solutions and lens cases.
Early Contact Lens Solutions
I began fitting contact lenses in the early 1970s when there were only a few solutions available for polymethylmethacrylate (PMMA) lenses. No FDA regulations for PMMA solutions and accessories existed. Soft lenses were originally approved under the drug regulations. The soft lens care system consisted of a homemade saline solution from distilled water and salt tablets. The device amendments weren't established until 1976. Gas Permeable (GP) lenses were introduced in the late 1970s and originally were packaged dry and shipped in flat pack cases that were sealed in pouches. Care systems for PMMA lenses consisted of whatever was available. Initially, these systems weren't regulated for compatibility with GP lenses.
Often, practitioners told patients they needed three different solutions to adequately care for PMMA lenses. Cleaners were too strong for lens storage, and disinfectant/storage solutions were inadequate for cleaning and too irritating to be used as solutions for insertion. Therefore, patients used three separate solutions for cleaning, storage/disinfection and insertion.
Soft lens solutions began as homemade saline followed by the introduction of weekly enzyme and surfactant cleaners. The lenses were thermally disinfected. This involved hanging the lens case in a modified baby bottle warmer and adding tap water to the heater to create steam disinfection. Later models were developed that were more like a heat sink than a vaporizer. Eventually, a unit was developed to plug directly into the wall, which looked similar to some of our modern-day cell phone chargers. The initial chemical soft lens disinfection system was a threebottle set from a company called Burton and Parsons, consisting of Normol (saline), Flexsol (the storage/disinfectant) and Preflex (the surfactant cleaner).
The soft lens saline solution made from salt tablets and 1-gallon jugs of easily contaminated distilled water was linked to a series of infections, the most serious were caused by Acanthamoeba. To hasten the elimination of homemade saline, the FDA approved preserved rinsing and storage solutions. These eventually led to the development of solutions that received a disinfection claim. Preservatives used in the initial chemical care systems often caused ocular irritation and red eyes, because of the degradation of the preservative thimerosal, as well as the uptake and release of the small molecular weight preservative Chlorhexidine from the lens. Patients failed to clean their cases and frequently topped off the solution remaining in the case from the previous night.
To reduce ocular irritation and allergic reactions from these chemicals, industry introduced peroxide systems for disinfection. In addition, industry introduced unit-dose nonpreserved saline, aerosol nonpreserved saline and sorbic acid preserved saline to decrease eye irritation upon lens application. The directive was to rinse the lenses with one of these neutral and supposedly nonirritating products. As with the chemical care systems, there was the possibility that residual peroxide would cause a problem. The sorbic acid preservatives caused some fairskinned patients to develop contact dermatitis with red streaks when the excess saline ran down their faces. Aerosol nonpreserved saline solutions were initially produced without buffers and were found to have a pH as low as 5.7, resulting in burning and stinging upon lens application. In addition, Acanthamoeba was found on the tips of the aerosol nozzles. The only reasonably comfortable products for lens application were the unit-dose buffered, nonpreserved saline solutions. Unfortunately, these were expensive, and people often kept the open vials for a few days, which raised the risk of contamination and infection.
FDA Clinical Controls
To ensure the safety and efficacy of investigational devices, the Investigational Device Exemption (IDE) was established to enforce controlled clinical trials. IDEs are granted in both significant and nonsignificant risk categories. Nonsignificant risk studies are performed under an abbreviated IDE, which must be approved by an Institutional Review Board (IRB) before the study is implemented. Nonsignificant risk studies include daily wear contact lenses and solutions with components previously used in approved products, while significant risk studies primarily are restricted to extended wear contact lenses and solutions with new components. Both an IRB and the FDA must review and approve significant risk study protocols before clinical trials begin. To ensure safe progress throughout the study, the FDA requires IDE progress reports to be sent to the IRB. The IRB and the study's sponsor are responsible for monitoring the trial.
Clinical investigations completed before market clearance, or approval, generally involve a small number of patients and qualified practitioners. Studies are extremely well organized and monitored. Following the clearance/approval of a device, the floodgates of noncompliance open widely. One study (Efron, 2004) indicated that more than 40 percent of patients are noncompliant in one or more aspects of contact lens care. Thus, the FDA has initiated Post Approval Surveillance (PAS) studies for certain significant risk products to monitor results after product marketing.
Device Classifications — The general history and potential risks/benefits of a product determine the class into which it's placed. (Class I general controls; Class II special controls; Class III premarket approval). Class I general controls are insufficient in ensuring the safety and efficacy of contact lenses and solutions.
Class II special controls are used to determine the substantial equivalency of investigational contact lenses or solutions compared with similar, previously approved products. Nonclinical and preclinical testing is performed on lenses and solutions. And if the results are satisfactory, clinical trials may begin (Table 1).
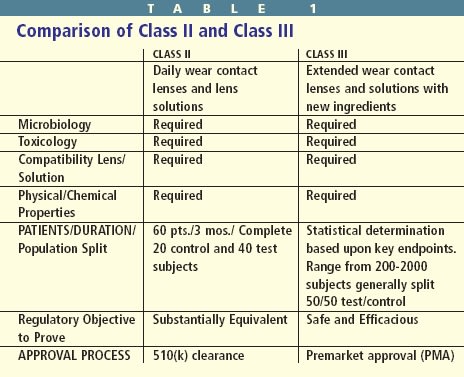
Class III applications also require appropriate nonclinical and preclinical testing. Class III contact lenses and care solutions must also demonstrate safety and efficacy. This requires a statistically justified population. These studies generally have a larger patient population and are conducted for a longer period to enhance the level of confidence that the study will demonstrate product safety and efficacy.
While state laws and licensure govern practitioners, the FDA regulates manufacturers. The FDA's approval terms for a given product are described in the approved labeling section that appears on and inside the packaging. With contact lens solutions, the labeling insert usually includes the purposes of the product, chemical descriptions, actions, use indications and contraindications, warnings and precautions, adverse reactions, directions for use, shelf-life and in some solutions, recommended discard dates. Unfortunately, many patients don't read the labeling insert. Although the FDA doesn't directly govern practitioners, prescribing or recommending devices in any way that differs from its original FDA labeling can lead to legal problems.
Guidance Documents
In 1976, the U.S. Congress enacted Medical Device Amendments to regulate medical devices differently from drugs, and the FDA established within its structure the Bureau of Medical Devices (now the Center for Devices and Radiological Health). Some medical devices, including contact lenses and solutions, that the Bureau of Drugs had regulated as new drugs were considered transitional. This placed them in Class III, the strictest device regulatory group, where they remained until some of them were reclassified to Class II in 1993.
Congress rewrote the Medical Device Amendments and passed a new law on Nov. 28, 1990. This Safe Medical Device Act contains provisions specific to contact lenses and their subsequent regulatory status. The intent was to ensure all transitional devices the Bureau of Drugs regulated before 1976 were reclassified to Class II, assuming that no additional concerns of their safety or efficacy arose in a two- to three-year period, and that there were no challenges to this reclassification. In 1993, daily wear soft and GP contact lenses were placed in Class II in accordance with the Safe Medical Device Act of 1990. In May 1994, the FDA provided new guidance to reflect this change, while contact lens solutions and accessories were addressed in the 1997 guidance document.
The rest of the world came on board with device regulations around the same time the FDA issued the 1994 and 1997 guidance documents. Europe, Canada and Japan were in the process of adopting a uniform set of testing documents and quality systems under the umbrella of the International Organization for Standardization (ISO) certification (CE). All companies around the world who meet the requirements for ISO certification can apply the CE mark to their products. This allows the company to distribute the products in all 27-member European countries without additional approvals. Before the adoption of these quality systems, each European country regulated solutions and lenses differently or not at all. Lenses generally had minimal restrictions, and regulations for solutions ran the gamut from drug approvals to no approvals. The formation of the European Community (EC) allowed for the adoption of a more consistent approach to testing and granting approvals for the member countries. The FDA has adopted many of the ISO contact lens and care system test procedures; however, it doesn't recognize the application of the CE mark for distribution. Canada and Japan also recognize parts of the system but require local approval for distribution.
Nonclinical/Preclinical Testing
Before the FDA permits clinical testing, the manufacturing of the device, as well as its physical and chemical characteristics and toxicology and microbiology must be evaluated in an acceptable manner. These nonclinical/preclinical procedures must demonstrate that human subjects can use a device safely in a clinical trial (Table 2).
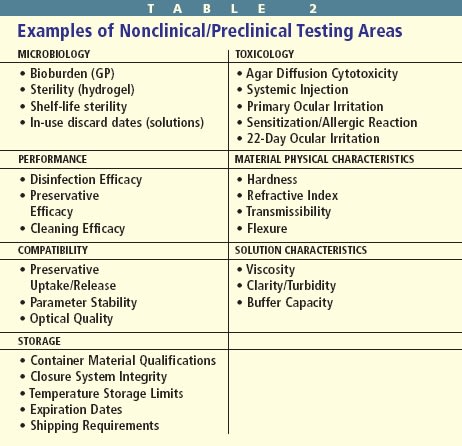
Microbiological testing is necessary, because hydrogel lenses and solutions require sterility, stability and shelf-life dating. Sterilization is the total elimination of a population of microorganisms. Regulations require manufacturers to deliver sterile contact lenses and solutions, and they must remain sterile until opened. Shelf-life dating indicates the duration the chemical integrity and sterility are maintained until the package is opened. Once containers are opened, contact lenses and solutions are immediately subject to external contamination. All multidose solutions must be able to maintain a certain level of preservative efficacy once the container is opened. This requirement applies to rewetting drops, cleaning solutions, rinsing solutions and multipurpose solutions.
Disinfection implies that a certain level of microorganisms have been killed, and that it's related to the product's indication for use with lenses outside of the bottle. Currently, there are two standards set to determine disinfection efficacy: the Stand-alone and Regimen criteria. In the Stand-alone test procedure, the solution is separately and independently inoculated with three bacteria and two fungi at a level of 106. The Stand-alone criterion requires at least a 3-log reduction for each of the bacterial challenges and at least a 1-log reduction for the molds and yeasts during the recommended soak times. Companies can use the Regimen test to evaluate solutions if they can't pass the Standalone test. The Regimen test involves inoculating lenses at the same level of 106. The companies then follow the label procedures for cleaning, rinsing and storage. To pass the Regimen test, a product must reduce the bacterial load by at least 1 log unit and have a total 5 log-unit reduction for all three bacteria and stasis for the fungi.
Data from a study (Efron, 2004) showed that significant contamination levels exist in many contact lens solutions and lens cases, and on used contact lenses. Patients who perform sterilization risk damaging their lenses. However, patients who disinfect their lenses by reducing the microbial load to an acceptable level help maintain the life of their lenses. Disinfection systems must be sufficiently bactericidal to accomplish this. Proper cleaning and rinsing before lens storage reduces the microbial burden by about 103. Chemicals in preservative-containing rinsing or lubricating solutions should be sufficiently bacteriostatic, so no new contaminants can grow in unopened containers. It's assumed that in opened containers the bacteriostatic effectiveness (preservative efficacy) will maintain contamination at safe levels during the expected period patients use the solution under normal conditions. However, it's been found that approximately 45 percent of opened and used solution bottles and accessories are contaminated.
Toxicology Analysis
What's just as important as disinfection is the issue of toxicological analysis. The chemical composition of contact lens materials and solutions makes toxicological analysis critical. Chemicals may be irritating or sensitizing to ocular tissues, and unpolymerized monomers of lens materials and color additives may produce toxic, leachable byproducts. Before researchers can test contact lens polymers, additives and solutions on human subjects, they must evaluate their potential toxicity in animal model, bench and laboratory testing. Only after these exhaustive and expensive tests substantiate sufficient safety can in vivo human studies begin. Nonhuman cytotoxicity tests include agar diffusion, primary ocular irritation, systemic injection, guinea pig sensitization and the 22-day ocular irritation rabbit test. Most tests involve complex protocols the scientific community accepts and modifies for ophthalmic application.
Recent Problems
The 1997 guidance document for contact lens solutions reduced the regulatory burden for solution care systems and included two pages on lens case approvals with one additional page for labeling requirements. There was no guidance or standard developed on the final lens case performance or on the interaction or compatibility of the lens case with the variety of solutions used for overnight storage and disinfection.
In the late 1990s, silicone hydrogel lenses were introduced. Similar to the initial debut of GP lenses 20 years earlier, no specific lens care solutions were designed for the new material even though silicone hydrogels presented with new surface properties due to their chemical composition, pore structure and surface treatments. Within a short time, articles appeared describing the chemical toxicity associated with solutions that were successfully used for years with hydrogel lenses. Certain solutions were considered to have a higher incidence of staining than others. Comparison grids were developed to look at the relationship between lenses and solutions. While these were interesting findings, arguments were made on both sides about their significance.
Suddenly in late 2005, reports began to surface about fungal infections in Singapore and Hong Kong, followed by similar infections in the United States. The infections were linked to the Renu with MoistureLoc formulation (Bausch & Lomb). No contamination due to product manufacturing was ever discovered. A complex series of factors was determined to be the cause of the infections, including the failure of patients to adequately clean their cases, the topping off of solution in the case from the night before and the absorption of the small molecular weight preservative Alexidine by the contact lenses and lens case. The practice of topping off solution and the absorption of the preservative by the lens and the case materials were thought to reduce the amount of preservative available to kill the organisms present in the lens case storage solution. Interestingly, for the past 25 years, patients who cared for soft lenses practiced the same poor hygiene as those who wore silicone hydrogels. These practices were linked to the fungal infections. In the end, no cause and effect concerning one element was ever proven, but the withdrawal of the product from the market and the resultant drop in fungal infections associated with contact lens wear indicates the solution was the major contributor to the outbreak.
Late in 2006, there were more reports of ocular infections associated with contact lens wear, and again the organism was Acanthamoeba. In May 2007, the Centers for Disease Control and Prevention (CDC) established a link between Complete MoisturePlus Solution (AMO) and Acanthamoeba infections that were occurring in the Chicago area. The main source of contamination was traced to the water supply and distribution in the area. While only a weak association was established, it was determined the product wasn't sufficiently effective against Acanthamoeba, and AMO recalled the product. Unfortunately, the rate of infection continued to climb in the Chicago area even after the removal of the product from the market. This would indicate the limited effectiveness of other care systems against Acanthamoeba.
Interestingly, the FDA has never approved of a testing method or established a standard for effectiveness against Acanthamoeba. Experts at a workshop (Microbiological Testing for Contact Lens Care Products, Jan. 22-23, 2009) sponsored by the FDA, the American Academy of Ophthalmology, the American Academy of Optometry, the American Optometric Association and the Contact Lens Association of Ophthalmologists, discussed various theories of the mechanism of Acanthamoeba infection, as well as approaches to creating an effective disinfectant. The hope is to first develop a standardized test method to determine the efficacy of a solution. Still, risk assessments must be completed to establish the level of effectiveness (short of complete kill) necessary during the disinfection process.
Causes of Problems
At the FDA Ophthalmic Device Panel Meeting in June 2008 and January 2009, experts from the FDA, CDC, industry and academia convened to discuss the current state of lenses, care systems and accessories (lens cases). Research was presented with various tests showing the effectiveness of care systems in a laboratory setting. For more than 20 years, the standardized testing had well served the FDA, industry and the end users. Infection rates were low and generally treatable without serious and long-lasting consequences. The sudden elevation in serious and difficult-to-treat conditions, such as fungal and Acanthamoeba infections, attracted the attention of not only the eyecare professions but also the news media and the legal system. The cause and effect of these infections weren�t easily defined, yet a call rang out for more stringent standards and uniform test procedures.
One of the problems plaguing the FDA and industry is the variety of conditions under which a contact lens care system is used. Some users are meticulous about lens replacement schedules, cleaning, rinsing, lens disinfection, and cleaning and replacing lens cases. Others are content to extend the life of the lens beyond the recommended replacement schedule, abbreviate or ignore the cleaning/rinsing steps and save money by topping off the solution in the case. Data from surveys indicate the vast majority of users fall into the second group. While these examples of the inappropriate use of lenses and care systems put the patient at a higher risk for infection, they aren't the only cause. Here are three others:
1. Environmental factors also play a role in the development of complications associated with lens wear. Patients aren't always diligent about washing their hands or handling the lenses in a clean environment. The bathroom, which is traditionally used for lens application and removal, isn't the ideal location for maximum cleanliness.
2. Lens cases have been linked to a number of problems with lens wear. Early soft lens cases were made with a hard plastic that could withstand repeated heat or steam disinfection. But even they would eventually crack or leak and harbor bacteria in the crevices. Originally, less expensive snap top cases were supplied with solutions to encourage frequent replacement. But users would keep them as spares and continue to use the old case until well after the snap top had broken and the lid would no longer seal.
3. Tap water has been associated with an increased risk for Acanthamoeba infections. Changes in environmental regulations for municipal water processing, as well as the decreased concentration and use of chemicals has caused an increase in the number of organisms in the water we drink. Tap water rinsing of GP lenses was common practice between using a concentrated cleaner and disinfecting solution for overnight storage. Over the past few years, references to tap water rinsing were removed from the labeling. It wasn't part of the care regimen for soft lenses, but it was suggested after cleaning and before air-drying the lens case. This approach, at least temporarily, reduced the microbial content of the case and removed old storage solutions. The current suggestions are to eliminate the tap water rinse/air drying and use multipurpose solutions to clean the case. There's even a suggestion that the bioburden would be reduced if the lens case were refilled with fresh solution during the day. While this might seem to be a good alternative, asking contact lens wearers to use an expensive multipurpose solution to clean and rinse their cases and then use additional solution during the day for case hygiene isn't realistic. Any suggestions made to change the way patients care for lens cases must be practical and affordable to encourage patient compliance.
Lens Case Compatibility
One of the issues concerning lens care systems is whether they're compatible with case materials. While cases are regulated as a class II medical device, the requirements for their approval have been independent of the solutions and lenses. Case designs and volume must be submitted to the FDA with a description of the material, physical and chemical data of the material and the name of the supplier. In addition, any colorants used must be water insoluble. Toxicity testing of the plastic involves systemic toxicology, acute ocular irritation and in vitro cytotoxicity measures. To date, there are no compatibility requirements for lens cases with the exception of those used in thermal disinfection, so they can withstand repeated exposure to heat.
Collaborative Effort
The FDA, industry, practitioners and patients will play a role in the successes or failures of the future contact lens market. There's little doubt the time has come to revisit the guidance, standards and testing procedures used in evaluating contact lenses and associated lens care products. The challenge is to develop inexpensive and reliable testing that can determine compatibility and predict clinical performance.
To improve patient compliance, practitioners must consistently use a combination of written, verbal and visual methods to reinforce proper contact lens use and care (Table 3). Some materials are approved for extended wear (up to 7 consecutive days and nights), and others are approved for continuous wear (up to 30 consecutive days and nights). The truth is some eyes can tolerate only daily wear regardless of the lens material.

As practitioners, we should remember the adage, "An ounce of prevention is worth a pound of cure." Helping patients from the outset to develop good lens use and lens care habits is much more effective than trying to detect and rectify bad habits later. CLS
Dr. Gleason is president of Foresight Regulatory Strategies, Inc., Wilmington, Mass. He's an experienced regulatory, quality and clinical affairs executive with a history of successful management in the United States and international medical device businesses.
To obtain the references for this article, please visit http://www.clspectrum.com/references.asp and click on document #168.