



Dystrophies of the corneal anterior limiting lamina (ALL), also known by the eponymous name Bowman’s layer, comprise less than 1% of corneal dystrophies.1 But, unlike many other corneal dystrophies, ALL dystrophies can be quite painful and require regular emergency visits to eyecare professionals for treatment of recurrent corneal erosions (RCE).
ALL dystrophy is commonly referred to as Reis-Bücklers dystrophy. But is there just one dystrophy of this corneal layer? Also of interest, how do Reis and Bücklers connect, as they published their descriptions of a dystrophy of the ALL three decades apart?
ALL dystrophy is a rare bilateral pathology that is inherited in an autosomal dominant pattern. It is commonly diagnosed in the first decade of life and usually advances to require corneal transplantation.2 This disease can reemerge in the donor tissue, but how does this reemergence occur? These are a few of the interesting obscurities that we will attempt to clarify in this article.
HISTORICAL PERSPECTIVE
The first description of a distinct anterior, subepithelial corneal dystrophy was offered by Reis in 1917 in Bonn, Germany, and was reported in Deutsche Medizinische Wochenschrift (German Medical Weekly Journal).3 Reis described this disease as a patchy loss of corneal transparency in a 20-year-old man, who disclosed that his father and three of his four siblings had the same anomaly. In this case, the corneal dystrophy was first diagnosed at age 5 years and was accompanied by prolonged periods of severe irritation in both eyes, which initially suggested herpes simplex keratitis. A lack of response to any treatment (not disclosed to Reis), however, led to the diagnosis of a corneal dystrophy.
Thirty-two years later, Bücklers added more details about this rare inherited dystrophy. He described four successive generations of one family, which, according to Duke-Elder and Leigh, may have originated from the family described by Reis.4,5 Bücklers’ account provided detailed illustrations, revealing cornea-wide clouding that followed a distinctive pattern, with areas of dense and less-dense transparency losses. While using an optical section with a biomicroscope, Bücklers also demonstrated that the focus of this corneal pathology is at the ALL level. Presumably, Bücklers’ more detailed, multi-page description with illustrations of the disease, published 32 years after Reis’ one-paragraph report, earned Bücklers the honor of joining Reis in a double eponym for this discovery.
MORE THAN ONE ALL DYSTROPHY?
Another 20 years passed, and the ALL dystrophy plot thickened. Grayson and Wilbrandt reported a dystrophy of the same corneal region as Reis-Bücklers, with a similar clinical manifestation that apparently was autosomal dominant.6 This dystrophy, termed the anterior membrane dystrophy of Grayson-Wilbrandt, is different in that it presents later in life, causes fewer erosions, and affects vision less. Grayson and Wilbrandt did not exclude the possibility of the disease being a milder variant of Reis-Bücklers dystrophy.
One year later, Thiel and Behnke also described an unknown, hereditary, subepithelial corneal dystrophy of the same corneal region.7 This disease is known as honeycomb dystrophy because of its clinical appearance.
Whether the Thiel-Behnke honeycomb-shaped dystrophy is a variant of Reis-Bücklers has been debated and, more recently, was the focus of a histopathological study of 28 corneas that had the clinical diagnosis of ALL dystrophy.8 The study authors concluded that Reis-Bücklers and Thiel-Behnke are two distinct and separate dystrophies of the ALL. Using light microscopy, the researchers described Thiel-Behnke as having band-shaped subepithelial deposits that stain positively with Masson trichrome, while ultrastructurally they reported “curly fibers.” The Thiel-Behnke variant resulted in total elimination of the ALL, which was replaced with undulating fibrocellular tissue. The basement membrane was frequently absent, and the presence of “curly” collagen was noted. The researchers further ventured that most cases reported in the literature as Reis-Bücklers were indeed Thiel-Behnke dystrophy.
Two studies reported that, ultrastructurally, the epithelium in Reis-Bücklers has variable thickness, with cystic and enlarged basal cells.8,9 Furthermore, they reported that the ALL had been lost and replaced by band-shaped deposits, which were also noted in the basal epithelium and the superficial stroma. Ultrastructurally, these deposits were electron-dense and rod-shaped, and they were associated with microfibrillar materials and large proteoglycan filaments.9
Like Reis-Bücklers, Thiel-Behnke corneal dystrophy is a rare bilateral disease characterized by anteriorly located gray opacities and progressive visual impairment. The disease is caused by a specific mutation in the transforming growth factor beta-induced gene and follows an autosomal dominant inheritance pattern.8,10
Based on histopathological ultrastructural reports by Küchle et al8 and by Liang et al,9 at least two distinctly different dystrophies are associated with the ALL. The differential diagnosis may be accomplished only by an ocular pathologist. The differential features are noted only histologically, at least in the earlier phase of the disease. Clinically, this differential diagnosis is not necessarily critical for ALL dystrophies. For example, when vision is affected sufficiently to interfere with how a patient functions in life, corneal transplantation must be considered, regardless of whether it is Thiel-Behnke or Reis-Bücklers dystrophy.
Typically, ALL dystrophies are bilateral, and they manifest at an early age. Initial clinical signs, which usually appear before the age of 10, include small, irregular opacities scattered throughout the central and midperipheral cornea.2 These opacities are usually axial and at the level of the ALL. As the dystrophy progresses, subepithelial opacities in the Thiel-Behnke form become a more identifiable honeycomb shape, with the peripheral cornea remaining uninvolved.11 This clinical distinction between Thiel-Behnke and Reis-Bücklers is not necessarily apparent in every case. As the disease progresses, the characteristic opacities progress deeper into the stromal layers, as is noted histologically. This further reduces visual acuity, while peripheral invasion may also be observed. The posterior stroma, posterior limiting lamina (Descemet’s layer), and endothelium are not affected.
DISEASE MANAGEMENT
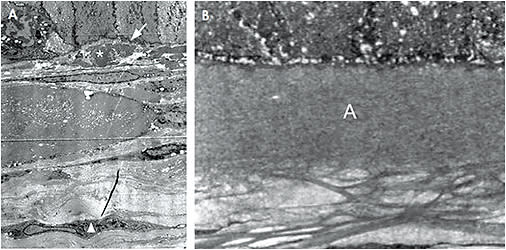
Dystrophies involving the ALL result in the loss of this corneal layer, to which the corneal epithelium is attached. This severely weakens the epithelial adhesion to the remainder of the cornea, causing frequent RCE episodes. Based on our clinical (Figure 1) and histopathological (Figure 2) observations of dystrophic corneas, including those that have ALL dystrophy, the RCE manifestation is specifically due to the elimination of type VII collagen fibers and of anchoring plaques, which are important components in epithelial adhesion to underlying connective tissue.12 Further compounding this epithelial fragility is the fact that the basement membrane produced by the epithelium is incompletely synthesized and exhibits frequent disruptions (Figure 3).
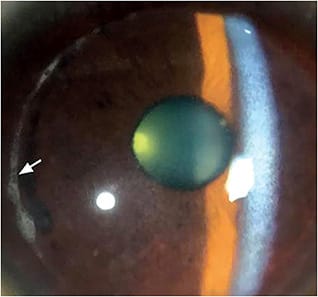

Patients who have ALL dystrophy will experience periods of moderate-to-severe ocular discomfort caused by intermittent RCEs, which require frequent visits to the office (Figure 4). These patients endure periodic episodes of ocular pain, foreign body sensation, photophobia, tearing, red eye, and spontaneous epithelial defects upon awakening.13 Fluorescein will stain the corneal epithelial irregularities, outlining areas of negative staining due to epithelial basement membrane damage.14
Treatment for acute episodes of ALL dystrophy that involve RCEs is similar to treatment for corneal abrasions (Table 1). Prescribe prophylactic topical antibiotics or ointments four to six times per day. Topical nonsteroidal anti-inflammatory drugs and cycloplegic drops may be prescribed up to three times per day to treat ocular discomfort and photophobia. It is important to follow up with patients every one to two days as the epithelium heals.
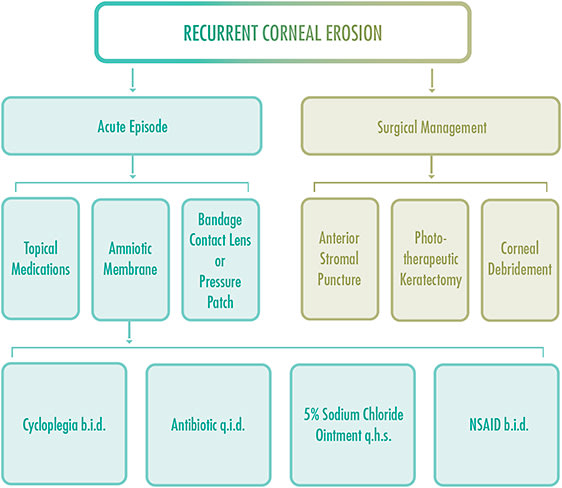
After re-epithelialization is complete, hypertonic saline ointment may be added to the treatment regimen for up to 12 months to reduce corneal edema and the risk for future RCEs. Patients may be monitored every three to six months, depending on the severity of the RCE. Persistent use of artificial tears, four to eight times per day, is important to maintain corneal lubrication.13
If a large epithelial defect is present, topical antibiotic drops and a pressure patch or bandage contact lens should be applied. Oral analgesics may be used as needed. Eye-care professionals should consider corneal debridement or a diamond burr polish for RCEs. Dehydrated or cryopreserved amniotic membranes may allow the epithelium to heal faster with minimal scarring.
Refer patients who experience multiple recurrences to a cornea specialist for surgical intervention, such as phototherapeutic keratectomy, anterior stromal puncture, superficial keratectomy, or neodymium-doped yttrium aluminum garnet (Nd:YAG) laser reinforcement to remove abnormal epithelium.13 This treatment may result in a small, visually insignificant corneal scar.
REEMERGENCE OF DISEASE
Since the early days of successful penetrating keratoplasty, clinicians have known that stromal dystrophies have a tendency to recur in patients who have undergone corneal transplantation.15 More recently, researchers have shown that dystrophies of the ALL have the highest frequency of recurrence among corneal connective tissue dystrophies.16 Therefore, it is important to counsel ALL dystrophy patients who need a transplant that the disease may reappear in the donor tissue and reverse the visual benefit achieved by the surgery.
This return of ALL dystrophy has been reported in keratoconus patients who had undergone penetrating or lamellar keratoplasty.17 Current keratoplasty surgery involves the removal of a 6mm-to-8mm corneal button from a host cornea that typically measures 10mm to 11mm.12 Therefore, it has been argued that keratoconus, which is a pancorneal disease, was never completely eliminated from the eye,17 allowing residual disease to invade the donor tissue. The latency interval for keratoconus appears to be one to two decades; but, for ALL dystrophy, the interval is just two years.16,17 The point has been raised that, as the disease was not completely removed, the correct terminology for the pathophysiology of this disease process is not “recurrence” but rather “reemergence.”
The clinical reemergence of the disease does not dictate an imminent need for a regraft. A second transplantation surgery may be indicated years later or not at all. Nevertheless, regular monitoring of post-transplant patients is good practice.
CURRENT UNDERSTANDINGS
We conclude here that there are two distinctly different dystrophies associated with the ALL: Thiel-Behnke and Reis-Bücklers. A third dystrophy reported by Grayson and Wilbrandt may exist or may be a variant of one of the other two. Nevertheless, Reis-Bücklers appears to be used as a global term for all ALL dystrophies. This may be because the clinical differentiation between Thiel-Behnke and Reis-Bücklers is often indistinct, and a final diagnosis may be possible only through histopathology. Interestingly, Thiel-Behnke appears to be the more common of the two established ALL dystrophies; thus, it is often mislabeled.
The ALL dystrophies lead to episodes of intermittent RCE syndrome and require frequent care from an eyecare professional. Patients who have ALL dystrophy are typically diagnosed within the first decade of life and, for this reason, will likely need a cornea transplant by the second decade. We also know that cornea transplant patients who have ALL dystrophy are at greater risk of reemergence of the disease than are patients who have other corneal dystrophies, and they need to be counseled about this possibility. CLS
The authors acknowledge with gratitude the experimental and intellectual contributions by Alan Burns, PhD. They also wish to recognize the generous and excellent translation of German texts by Ms. Lina Bienefeld.
REFERENCES
- Musch DC, Niziol LM, Stein JD, Kamyar RM, Sugar A. Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci. 2011 Sep;52:6959-6963.
- Kobayashi A, Sugiyama K. In vivo laser confocal microscopy findings for Bowman’s layer dystrophies (Thiel-Behnke and Reis-Bücklers corneal dystrophies). Ophthalmology. 2007 Jan;114:69-75.
- Reis W. Fämiliare, Fleckige Hornhautentartung. Dtsch Med Wochenschr. 1917;43:575.
- Bücklers M. Über eine weitere familiäre Hornhautdystrophie (Reis). Klin Monatsbl Augenheilkd. 1949;114:386-397.
- Duke-Elder S, Leigh AG. Disease of the Outer Eye: Corneal Dystrophies. In: System of Ophthalmology. Vol 8. Great Britain: Henry Kimpton Publishers; 1965:924-926.
- Grayson M, Wilbrandt H. Dystrophy of the anterior limiting membrane of the cornea. (Reis-Bücklers type). Am J Ophthalmol. 1966 Feb;63:345-349.
- Thiel HJ, Behnke H. Eine bisher unbekannte subepitheliale hereditäre Hornhautdystrophie [A hitherto unknown subepithelial hereditary corneal dystrophy]. Klin Monbl Augenheilkd. 1967;150(6):862-874.
- Küchle M, Green WR, Völker HE, Barraquer J. Reevaluation of corneal dystrophies of Bowman’s layer and the anterior stroma (Reis-Bücklers and Thiel-Behnke types): A light and electron microscopic study of eight corneas and a review of the literature. Cornea. 1995 Jul;14:333-354.
- Liang Q, Pan Z, Sun X, Baudouin C, Labbé A. Reis-Bücklers corneal dystrophy: a reappraisal using in vivo and ex vivo imaging techniques. Ophthalmic Res. 2014 May;51:187-195.
- Charukamnoetkanok P, Tuli S, Azar DT. Anterior Corneal Dystrophies: Dystrophies of the Epithelium, Epithelial Basement Membrane, and Bowman’s Layer. In: Smolin and Thoft’s The Cornea. Scientific Foundations & Clinical Practice. 4th ed. Foster S, Azar D, Dohlman CH, eds. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:813-824.
- Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015 Feb;34:117-159.
- Bergmanson JPG. Cornea. In: Clinical Ocular Anatomy and Physiology. 28th ed. Bergmanson JPG, ed. Houston: Texas Eye Research and Technology Center University of Houston College of Optometry; 2021:78-113.
- Friedman NJ, Kaiser PK, Pineda R. The Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology. 5th ed. Gabbedy R, Davie B, eds. Boston, MA: Saunders Elsevier; 2021:182.
- The Wills Eye Manual. 7th ed. Begheri N, Wajda B, eds. New York: Wolters Kluwer; 2017:54-55.
- Morgan G. Macular dystrophy of the cornea. Br J Ophthalmol. 1966 Feb;50:57-67.
- Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003 Jan;22:19-21.
- Bergmanson JP, Goosey JD, Patel CK, Mathew JH. Recurrence or re-emergence of keratoconus—what is the evidence telling us? Literature review and two case reports. Ocul Surf. 2014 Oct;12:267-272.